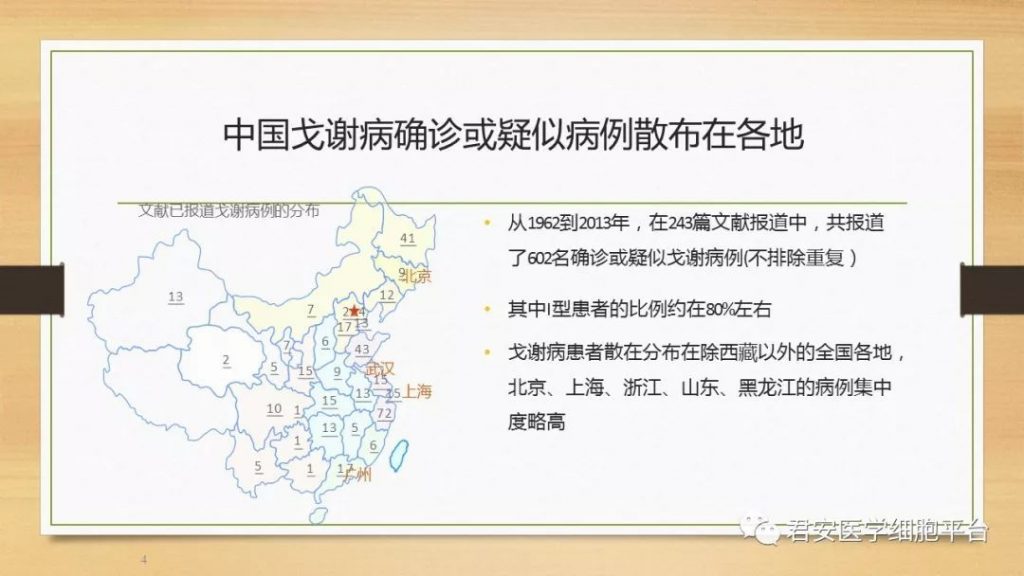
I型戈谢氏病患者的临床表现各不相同,个体间的症状与体征差别很大。这些症状和体征主要集中在以下4个解剖单位:内脏、血液系统、骨骼和代谢系统。这些症状与体征也常常出现于其他疾病,因此,医生们在诊断上往往不会马上考虑戈谢氏病,导致戈谢氏病患者出现症状到最后被确诊,其间通常需要一个较长的时间段。


确诊戈谢氏病的检查
对症状和体征的评估可以让医生对戈谢氏病有个初步的诊断,但需要通过几个检查进行确诊。过去,侵入性的骨穿最常应用。现在,可通过无损伤的检查来测定患者的酶水平进行确诊。低于或等于30%正常酶水平即可确诊。可测量白细胞或成纤维细胞的酶活性。
酶学测定
当葡糖脑苷脂酶活性为正常值的30%以下时,即可确诊戈谢氏病 。
酶学检测比骨穿更精确,并且更少损伤。
DNA 检测
DNA 检测可用于犹太民族的戈谢氏病诊断,有4种特殊的基因变异占了犹太戈谢氏病患者的89% ~ 96%。当用于普通人群的诊断时,这种方法没有酶学检测灵敏。
DNA检测也可以帮助明确携带者。
其他评估手段
除外酶学及基因检测,也可用其他方法对戈谢氏病进行评估,如骨骼成像,血液学分析、脏器评估等。点击监测指南,可以了解更多辅助检查方法。
诊断戈谢氏病
戈谢氏病的诊断对医学专家而言一直是个不小的挑战,尽管现在已经有了非常简便的诊断方法(酶学检测及基因检测)。误诊并不少见,常导致患者不能及时治疗并获得最佳的治疗效果。
诊断困难部分原因是因为疾病的罕见性,症状和体征没有特异性,特别是很多症状可以存在于其他疾病,因此临床医生不会马上考虑到戈谢氏病的可能性而让患者做相应的检查。而且,戈谢氏病的临床进展不能预测,有些症状和体征需要进展多年才被发现。
一些常见的误诊
戈谢氏病的许多症状和体征与其他疾病相似,因此很容易被误诊。常被诊断为以下疾病:
白血病
淋巴瘤
出血性疾病
骨髓炎
莱格-卡尔维-佩尔斯特三氏症候群
同时,有些疾病也有增大的细胞,与戈谢氏细胞相像,故会被误诊为戈谢氏病。在以下情况会出现“假戈谢氏细胞”
慢性粒细胞性白血病
多发骨髓瘤
何杰金病
I型戈谢氏病疾病进展
疾病进展
I型戈谢氏病的病程是不断进展的,可发展为很难或者根本不能逆转的病变。个体间的疾病进展很不相同并不能预测。在某些病例,戈谢氏病是致命的。患者常因硬化、脓毒血症、肺病以及术后并发症等引起死亡。
体格改变
长期的体格改变包括:
脾脏增大,脾梗死,疤痕和结节
进展性肝脏病变或门脉高压
进展性骨病变,骨坏死和永久性残疾
肺动脉高压
生活质量改变
戈谢氏病会严重影响患者的身体健康及生活质量,妨碍患者参与正常的生活、学习和工作, 影响家庭乃至社会的和谐。
遗传与戈谢氏病

戈谢氏病是一种常染色体隐性遗传疾病,因位于1号染色体长臂上的葡糖脑苷脂酶基因的突变引起。目前为止,已明确有超过300多种变异基因,这些变异会引起酶的稳定性下降,或结合能力下降。有相同基因型的患者,个体之间的临床表现差异很大。
犹太人群的发病率
尽管戈谢氏病可以发生在任何种族人群,但东欧(犹太人群)的发病率相对更高(850人中间可有一个), 而其携带者的比率可为12~15人中间有一个。 4种最常见的变异类型占到犹太人群中变异类型的 89% ~ 96%。
戈谢氏病的遗传概率
如果父母一方为戈谢氏病基因的携带者,则每一次妊娠,有50%的概率会把变异基因传给下一代。如果父母双方均为携带者,则每一次怀孕,孩子患戈谢氏病的几率为25%。
遗传咨询与检测
高危人群应进行遗传咨询和DNA 检测来除外戈谢氏病。高危人群包括:戈谢氏病患者的亲属,犹太后裔以及有症状的个体。
文章属于病友个人治疗见解及自身对疾病的治疗分析仅供参考(不代表本站同意其说法)谨慎参阅!
提示:ITP6病友网站内任何对疾病的建议都不能替代执业医师当面诊断!
作者:血小板减少交流群病友分享(文章内图片于群内交流)
链接:https://www.itp6.com/12131.html
文章版权归作者所有,未经允许请勿转载。
 微信扫一扫
微信扫一扫 